

The encoding and decoding of genetic information inevitably causes damage to its genomic DNA "vector". Multiple cellular DNA repair pathways work together to maintain the stability and integrity of the genome. However, as every coin has two sides, programmed DNA lesions can occur endogenously in certain cell types, which can be further channeled into altered genetic coding in the form of mutation, recombination, etc., thus allowing cells to acquire new functions. In this way, cells can acquire new functions. The diversification of antigen receptor genes is an example of a process initiated by programmed DNA lesions.
The Meng lab studies antibody diversification in B cells at the intersection of B cell biology and DNA repair. The lab applies immunology, biochemistry, functional-omics, and synthetic biology approaches and uses animal models and human samples to study the molecular mechanisms of cytidine deaminase AID targeting and error-prone decoding of programmed DNA lesions in B cells. Meanwhile, we also aim to develop platform technologies for therapeutic antibody discovery based on immune diversity studies. Along the way, we have revealed common and unique features of programmed DNA lesion repair in B cells, proposed new theories of AID targeting at different scales under either physiological or pathological conditions, and established platform technologies for ex vivo antibody diversification. Our research focuses on the following areas:
(1) The role of cis- and trans-factors, including DNA sequence, chromatin features, and DNA metabolism factors, in facilitating robust antibody diversity;
(2) Methods to unleash the power of programmed DNA lesions in antibody discovery, cancer immunity, gene editing, etc.
We found transcription core factor ELOF1 can stabilize RNA Polymerase II on chromatin to support different transcription-coupled DNA metabolic processes. (Dai et al. Mol Cell, 2025)
 ELOF1 facilitates transcription-coupled AID targeting and DNA repair.In adaptive immunity, transcription-coupled damage is introduced into antibody genes by activation-induced cytidine deaminase (AID) to diversify antibody repertoire. However, the coordination between transcription and DNA damage/repair remains elusive. Here, we find that transcription elongation factor 1 (ELOF1) stabilizes paused RNA polymerase II (RNAPII) at transcription barriers, providing a platform for transcription-coupled DNA damage/repair. Using a genetic screen, we discover that ELOF1 is required for AID targeting and that ELOF1 deficiency results in defective antibody class switch recombination and somatic hypermutation in mice. While downstream transcription-coupled repair factors are dispensable for AID-damage, ELOF1 mechanistically facilitates both transcription-coupled damage and repair by stabilizing chromatin-bound RNAPII. In ELOF1-deficient cells, paused RNAPII tends to detach from chromatin and fails to recruit factors to induce or repair DNA damage. Our study places ELOF1 at the center of transcription-coupled DNA metabolism processes and suggests a transition of RNAPII from elongation to a DNA damage/repair scaffold.
ELOF1 facilitates transcription-coupled AID targeting and DNA repair.In adaptive immunity, transcription-coupled damage is introduced into antibody genes by activation-induced cytidine deaminase (AID) to diversify antibody repertoire. However, the coordination between transcription and DNA damage/repair remains elusive. Here, we find that transcription elongation factor 1 (ELOF1) stabilizes paused RNA polymerase II (RNAPII) at transcription barriers, providing a platform for transcription-coupled DNA damage/repair. Using a genetic screen, we discover that ELOF1 is required for AID targeting and that ELOF1 deficiency results in defective antibody class switch recombination and somatic hypermutation in mice. While downstream transcription-coupled repair factors are dispensable for AID-damage, ELOF1 mechanistically facilitates both transcription-coupled damage and repair by stabilizing chromatin-bound RNAPII. In ELOF1-deficient cells, paused RNAPII tends to detach from chromatin and fails to recruit factors to induce or repair DNA damage. Our study places ELOF1 at the center of transcription-coupled DNA metabolism processes and suggests a transition of RNAPII from elongation to a DNA damage/repair scaffold.
We uncovered an unexpected non-coding role of the antibody coding sequence in AID targeting through regulation of DNA flexibility. (Wang et al., Cell, 2023)
 Non-coding regulation of AID by the Ig coding sequence on the nm scale. In the antibody variable region, complementarity determining regions (CDRs) interact directly with antigenic epitopes, while framework regions (FRs) maintain the antibody protein structure. In the exon (~350bp), AID preferentially targets CDR sequences and generates mutation hotspots. Although this phenomenon is widely recognized, the mechanism of CDR bias is less understood. By combining AID biochemical assays and several mouse models, we found that the DNA sequence per se determines AID targeting activity. Analysis of AID enzymatic activity on more than 1,000 IgV sequences from 25 species revealed that the amino acid coding sequences of CDRs are more favorable substrates for AID. Furthermore, through single-molecule and in silico simulation experiments, we found that AID tends to act on DNA sequences with higher flexibility. With the revealed molecular mechanism, we can artificially generate AID hotspot in vivo and ex vivo. The paper presents mesoscale sequence cues for somatic hypermutation to explain why the process of antibody diversification is focused on the three complementary determining regions (CDRs).
Non-coding regulation of AID by the Ig coding sequence on the nm scale. In the antibody variable region, complementarity determining regions (CDRs) interact directly with antigenic epitopes, while framework regions (FRs) maintain the antibody protein structure. In the exon (~350bp), AID preferentially targets CDR sequences and generates mutation hotspots. Although this phenomenon is widely recognized, the mechanism of CDR bias is less understood. By combining AID biochemical assays and several mouse models, we found that the DNA sequence per se determines AID targeting activity. Analysis of AID enzymatic activity on more than 1,000 IgV sequences from 25 species revealed that the amino acid coding sequences of CDRs are more favorable substrates for AID. Furthermore, through single-molecule and in silico simulation experiments, we found that AID tends to act on DNA sequences with higher flexibility. With the revealed molecular mechanism, we can artificially generate AID hotspot in vivo and ex vivo. The paper presents mesoscale sequence cues for somatic hypermutation to explain why the process of antibody diversification is focused on the three complementary determining regions (CDRs).
We identified a novel DNA double-strand break (DSB) repair protein, ERCC6L2, in deletional DNA recombination during antibody class switch recombination. (Liu et al., Cell Res, 2020)
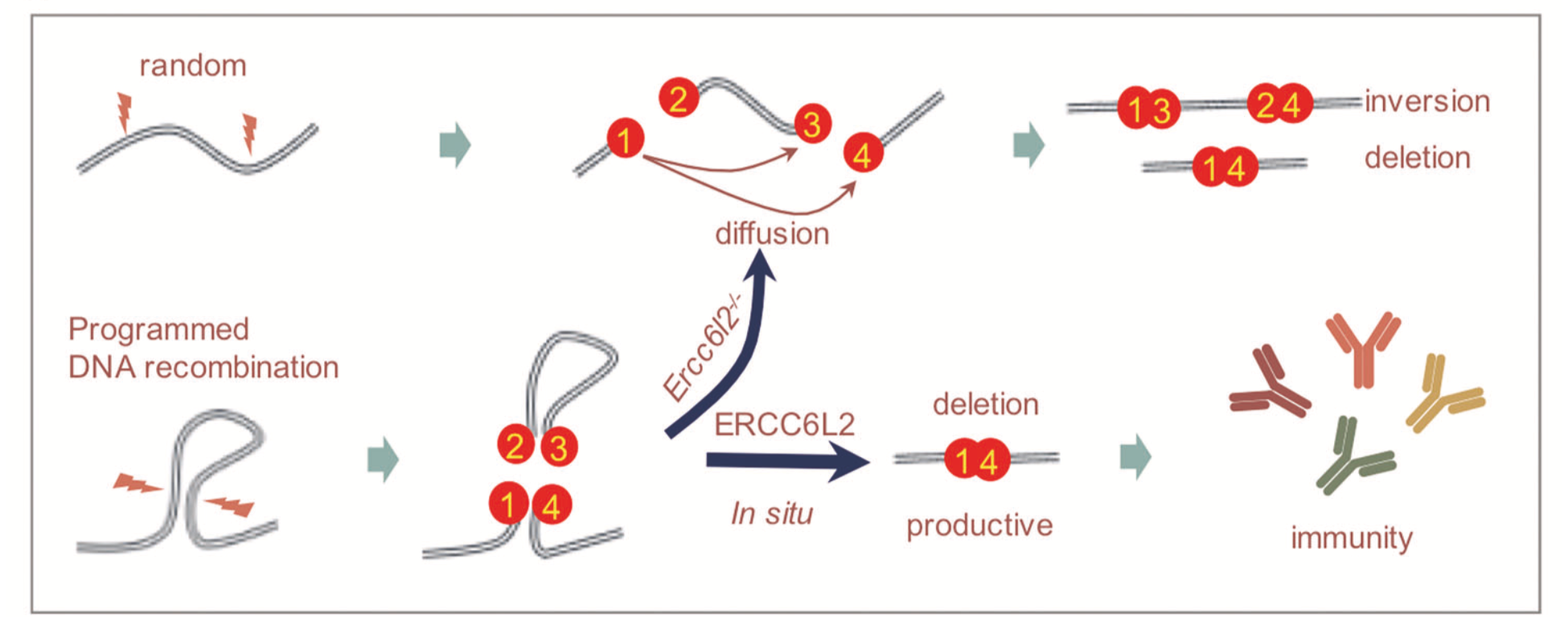 ERCC6L2 facilitates orientation-specific end joining in CSR. In antibody class switch recombination (CSR), the two DSBs initiated by AID occurred tandemly in the IgH constant region. DNA end joining in this process is orientation specific, i.e. most DNA recombination occurs in the manner of "intermediate fragment deletion", ensuring that most recombinant gene products can successfully encode antibody genes. We combined CRISPR and chemical genetic screening methods to construct a genetic map of DNA damage repair in B cells. In this map, we discovered a novel non-homologous end-joining (NHEJ) protein, ERCC6L2. ERCC6L2 deficiency results in defective DSB repair and immunodeficiency. Using cellular and mouse models, we demonstrated that ERCC6L2 is directly involved in DSB end joining and further uncovered its critical role in orientation-specific recombination. Using a genetically modified mouse model, we also demonstrated that the potential catalytic activity of ERCC6L2 is required for deletional CSR.
ERCC6L2 facilitates orientation-specific end joining in CSR. In antibody class switch recombination (CSR), the two DSBs initiated by AID occurred tandemly in the IgH constant region. DNA end joining in this process is orientation specific, i.e. most DNA recombination occurs in the manner of "intermediate fragment deletion", ensuring that most recombinant gene products can successfully encode antibody genes. We combined CRISPR and chemical genetic screening methods to construct a genetic map of DNA damage repair in B cells. In this map, we discovered a novel non-homologous end-joining (NHEJ) protein, ERCC6L2. ERCC6L2 deficiency results in defective DSB repair and immunodeficiency. Using cellular and mouse models, we demonstrated that ERCC6L2 is directly involved in DSB end joining and further uncovered its critical role in orientation-specific recombination. Using a genetically modified mouse model, we also demonstrated that the potential catalytic activity of ERCC6L2 is required for deletional CSR.
We found that C>G base editors(CGBE) would generate a significant number of DNA double-strand breaks (DSBs) and developed new CGBEs by incorporating the DNA damage repair (DDR) factor HMCES. (Huang et al., Nat Cell Biol, 2024)
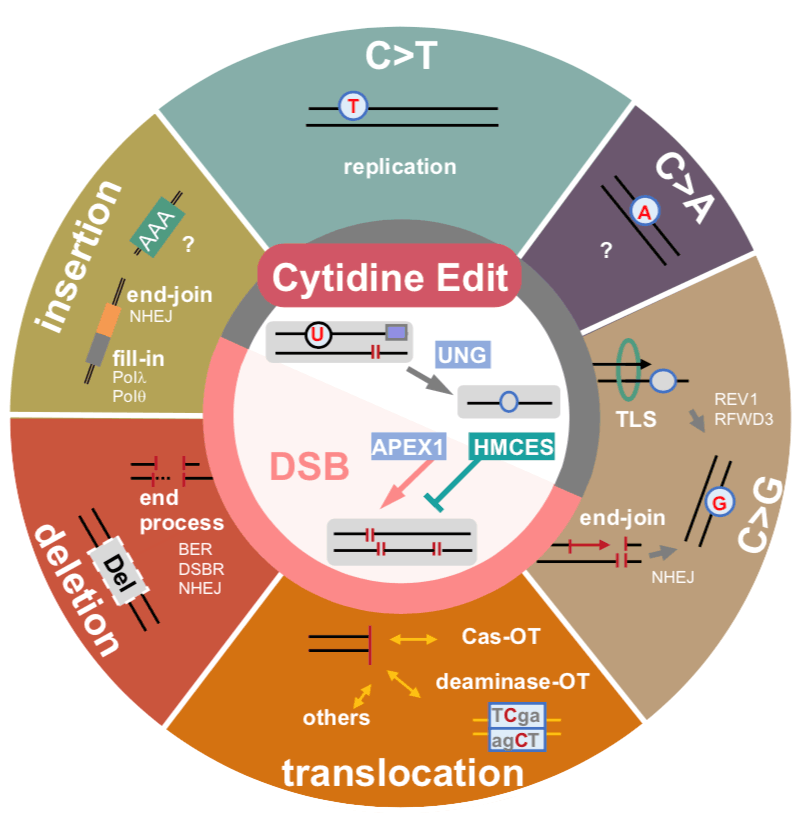
DSBs are the main source of CGBE’s by-products. Base editors (BEs) introduce base substitutions without double-strand DNA cleavage. Besides precise substitutions, BEs generate low-frequency ‘stochastic’ byproducts through unclear mechanisms. We performed in-depth outcome profiling and genetic dissection, revealing that C-to-G BEs (CGBEs) generate substantial amounts of intermediate double-strand breaks (DSBs), which are at the centre of several byproducts. Imperfect DSB end-joining leads to small deletions via end-resection, templated insertions or aberrant transversions during end fill-in. Chromosomal translocations were detected between the editing target and off-targets of Cas9/deaminase origin. Genetic screenings of DNA repair factors disclosed a central role of abasic site processing in DSB formation. Shielding of abasic sites by the suicide enzyme HMCES reduced CGBE-initiated DSBs, providing an effective way to minimize DSB-triggered events without affecting substitutions. This work demonstrates that CGBEs can initiate deleterious intermediate DSBs and therefore require careful consideration for therapeutic applications, and that HMCES-aided CGBEs hold promise as safer tools.